
Programme de la formation en ligne de la pharmacovigilance des essais cliniques
La formation en ligne « La pharmacovigilance des essais cliniques pour les RMR » en format elearning permet de prendre conscience du rôle du RMR (Responsable Médical Régional) dans la pharmacovigilance des essais cliniques et de connaître les différentes étapes de sa mise en œuvre tout en étant au fait des dernières évolutions de la réglementation.
![]()
Objectif pédagogique
Connaître la réglementation et les bonnes pratiques des essais cliniques.
Comprendre le suivi et la gestion de la pharmacovigilance des essais cliniques.
Cerner l’implication du RMR dans la pharmacovigilance des essais cliniques.
![]()
Pour qui ?
Référents médicaux en région (RMR/MSL), et autres collaborateurs en lien avec les services affaires médicales.
![]()
Format E-Learning
- Tout à distance, en complément de vos stages présentiels, gamifiés ou en immersion, nos produits sont conçus dans des formes pédagogiques innovantes et interactives.
- Le contenu est animé et scénarisé, intégrant QCM et validation des acquis. L’utilisateur accède à la formation à partir d’un simple navigateur (Chrome, Firefox, Edge, Safari, …).
En savoir plus sur la formation…
Durée
30 minutes
Tarif
100 € HT par apprenant + coût d’accès à la plateforme.

Besoin de personnaliser cette formation ?
Cette formation peut être personnalisée en fonction de vos objectifs et des spécificités de votre entreprise. Nous sommes à votre disposition pour créer un module sur-mesure ou compléter le module existant.
En savoir plus sur la personnalisation et nos formations sur-mesure…
La formation en ligne « La pharmacovigilance des essais cliniques » – Formation en ligne elearning est disponible en format PDF.
TOUTES NOS FORMATIONS EN LIGNE| E-LEARNING DE LA THÉMATIQUE
Formations affaires médicales
IFIS Interactive vous propose des formations e-learning en phase avec l’évolution de la réglementation et de la demande des industriels.
Les + de la formation en ligne « La pharmacovigilance des essais cliniques »
Vous prendrez conscience du rôle du RMR dans la pharmacovigilance des essais cliniques.
Vous connaîtrez les différentes étapes de la mise en œuvre de la pharmacovigilance.
Vous serez au fait des dernières évolutions de la réglementation.
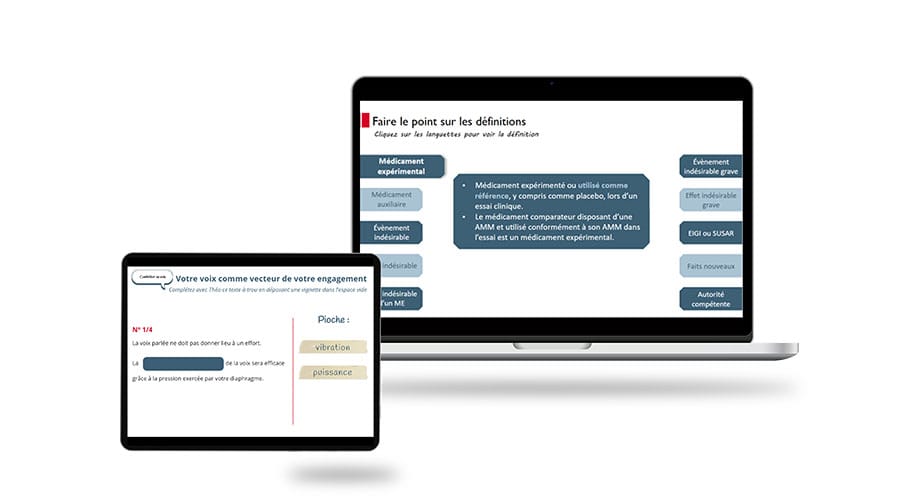
Programme détaillé de la formation en ligne « Pharmacovigilance des essais cliniques »
Objectifs pédagogiques
Connaître la réglementation et les bonnes pratiques des essais cliniques.
Comprendre le suivi et la gestion de la pharmacovigilance des essais cliniques.
Cerner l’implication du RMR dans la pharmacovigilance des essais cliniques.
Date de modification du produit : 02/2026

Programme de la formation pharmacovigilance des essais cliniques
La réglementation et les bonnes pratiques des essais cliniques
- Cadre réglementaire européen et français
- Rôle et responsabilité des différents acteurs (investigateur, promoteur, moniteur, ARC, pharmacovigilant, CRO, …)
Le suivi et la gestion des essais cliniques du point de vue de la pharmacovigilance
- Le protocole
- La brochure pour l’investigateur
- Le cahier d’observation
- Le suivi d’un essai clinique
- La gestion des cas
- La réconciliation des bases de donnée
- Le management du signal
- Les rapports (DSURs, PSUR/PBRER, plan de gestion du risque)
- La qualité
Le rôle et les missions du RMR dans la pharmacovigilance des essais cliniques
- Rôle et missions propres au RMR dans la pharmacovigilance
- Interface avec les différents acteurs
Format E-learning
Pédagogie
Modalité e-learning format standard. Document construit pédagogiquement. Le contenu est animé, scénarisé ainsi qu’une médiatisation dite standard. Utilisation de l’outil Storyline.
L’e-learning, ou formation à distance, permet de dispenser des programmes via une plateforme numérique interactive.
Il permet ainsi aux apprenants de :
- Suivre la formation au moment et dans le lieu qui leur conviennent le mieux
- Revenir à leur guise sur des points importants, mal compris ou mal assimilés
- Vérifier leurs connaissances grâce à des questionnaires d’évaluation
- Télécharger leur attestation d’assiduité
Équipement et spécifications recommandées
- Systèmes d’exploitation (a minima) : Windows 10
- Processeur (a minima) : Pentium Core I5
- Mémoire (a minima) : 4 à 8 Go en fonction du système
- Navigateurs : Edge, FireFox, Chrome (v88 et suivant), Safari (v9 ou suivant)
- Activer les cookies de session sur les postes utilisateurs
- La formation est adaptée aux formats mobile et tablette
TAUX DE SATISFACTION :
86,6%
Informations complémentaires
Public concerné
Référents médicaux en région (RMR/MSL), et autres collaborateurs en lien avec les services affaires médicales.
Prérequis
AUCUN.
Délai d’accès à la formation e-learning
10 jours à réception du fichier d’inscription.
Détail tarification
- Coût d’accès forfaitaire de 499 € HT ouvrant les droits d’accès à notre plateforme de formation pour 5 ans.
- + Coût annuel de connexion nominative à la formation : 100 € HT par salarié apprenant.
Personnalisation de nos modules e-learning et formations en ligne sur mesure
Personnalisez votre E-formation
Cette formation peut être personnalisée au vu des attentes particulières et/ou des spécificités de votre entreprise.
Évaluation par quiz de type QCM pouvant être effectué post-formation sur plateforme informatique. A l’issue de l’évaluation des acquis, les résultats sont communiqués.
Les formations e-learning d’IFIS Interactive sont aussi accessibles aux personnes en situation de handicap.
Nous sommes à votre écoute pour étudier ensemble les modalités d’accès requises.

Vos besoins sont spécifiques ?
Découvrez nos formations sur-mesure !
Nous innovons en vous proposant des formations sur-mesure pensées pour l’apprenant. Notre but : accompagner vos collaborateurs vers la réussite en personnalisant de A à Z votre solution e-learning pour les secteurs pharmaceutique, cosmétique ou du dispositif médical.
Foire aux questions
Que couvre la formation Pharmacovigilance des essais cliniques ?
Elle permet de comprendre le rôle du RMR dans la pharmacovigilance des essais cliniques et de connaître les étapes de sa mise en œuvre.
À qui s’adresse cette formation ?
Elle vise les référents médicaux en région, RMR/MSL, et les collaborateurs en lien avec les services affaires médicales.
Quel rôle joue la pharmacovigilance dans les essais cliniques du médicament à usage humains?
La pharmacovigilance joue un rôle crucial dans les essais cliniques, qui sont des études menées pour évaluer l’innocuité et l’efficacité d’un médicament ou d’un dispositif médical avant qu’il ne soit mis sur le marché. L’Agence nationale de sécurité du médicament et des produits de santé (ANSM) est l’autorité de réglementation en France chargée de superviser et de réglementer les essais cliniques.
La pharmacovigilance a plusieurs rôles à jouer dans les essais cliniques :
- Assurer la sécurité des patients : La pharmacovigilance a pour objectif de protéger la santé des patients en veillant à ce que les médicaments testés soient sûrs et efficaces. Cela implique de recueillir et d’analyser les données de pharmacovigilance pendant l’essai clinique, afin de détecter rapidement tout effet indésirable grave ou inattendu.
- Rédiger le protocole de l’essai clinique : Le protocole est le document qui décrit le déroulement de l’essai clinique, les objectifs, les populations étudiées, les critères d’inclusion et d’exclusion, et les procédures de suivi. La pharmacovigilance est impliquée dans la rédaction du protocole afin de s’assurer que les mesures de sécurité et les critères de suivi sont adéquats et conformes aux réglementations en vigueur.
- Evaluer les données de sécurité : La pharmacovigilance est chargée d’évaluer les données de sécurité recueillies pendant l’essai clinique, afin de déterminer si le médicament est sûr et efficace. Cela peut nécessiter l’analyse de données sur les effets indésirables, les résultats cliniques, et l’utilisation du médicament dans des populations spécifiques.
- Rédiger le rapport final de l’essai clinique : Le rapport final de l’essai clinique est un document qui présente les résultats de l’essai et une évaluation du profil bénéfice-risque du médicament. La pharmacovigilance est chargée de rédiger cette évaluation en se basant sur les données de sécurité recueillies pendant l’essai.
En résumé, la pharmacovigilance a un rôle central dans les essais cliniques, en veillant à la sécurité des patients et en évaluant l’innocuité et l’efficacité des médicaments testés.
Quelle est la définition des essais cliniques selon l'OMS ?
Selon l’Organisation mondiale de la santé (OMS), les essais cliniques sont des études scientifiques menées pour évaluer l’efficacité et la sécurité de médicaments, de vaccins, de dispositifs médicaux ou de traitements médicaux. Ils sont menés sur des personnes volontaires (appelées « sujets d’essai ») et sont conçus pour répondre à des questions précises sur l’efficacité et la sécurité d’un traitement.
Il existe plusieurs types d’essais cliniques, qui varient en fonction de l’objectif de l’étude et de la population cible. Par exemple, les essais cliniques de phase I sont principalement destinés à évaluer la sécurité d’un traitement et sont menés sur un petit nombre de sujets en bonne santé. Les essais cliniques de phase II et III sont destinés à évaluer l’efficacité d’un traitement et sont menés sur un plus grand nombre de sujets, y compris des personnes souffrant de la maladie ou du problème de santé ciblé par l’étude. Les essais cliniques de phase IV sont menés après l’autorisation de mise sur le marché d’un médicament et visent à évaluer l’efficacité et la sécurité du médicament dans la pratique clinique courante.
En résumé, les essais cliniques sont des études scientifiques menées pour évaluer l’efficacité et la sécurité de médicaments, de vaccins, de dispositifs médicaux ou de traitements médicaux sur des personnes volontaires. Ils sont menés selon une progression en différentes phases et visent à répondre à des questions précises sur l’efficacité et la sécurité des traitements étudiés.
Quels sont les différents rapports émis dans le cadre de la pharmacovigilance des essais cliniques ?
Dans le cadre de la pharmacovigilance des essais cliniques, il existe plusieurs types de rapports qui peuvent être émis :
- Les rapports d’événements indésirables (EI) : il s’agit de rapports qui signalent tout effet indésirable observé chez un sujet d’essai lors d’un essai clinique. Ces rapports doivent être envoyés au promoteur de l’essai et à l’organisme de réglementation compétent.
- Les rapports d’incidents graves : il s’agit de rapports qui signalent tout incident grave observé chez un sujet d’essai lors d’un essai clinique. Ces rapports doivent être envoyés au promoteur de l’essai et à l’organisme de réglementation compétent.
- Les rapports d’incidents graves imputables au médicament d’essai : il s’agit de rapports qui signalent tout incident grave imputable au médicament d’essai observé chez un sujet d’essai lors d’un essai clinique. Ces rapports doivent être envoyés au promoteur de l’essai et à l’organisme de réglementation compétent.
- Les rapports d’incidents graves imputables au médicament d’essai avec une issue fatale : il s’agit de rapports qui signalent tout incident grave imputable au médicament d’essai ayant entraîné une issue fatale chez un sujet d’essai lors d’un essai clinique. Ces rapports doivent être envoyés au promoteur de l’essai et à l’organisme de réglementation compétent.
- Les rapports d’incidents graves imputables au médicament d’essai avec une issue grave non fatale : il s’agit de rapports qui signalent tout incident grave imputable au médicament d’essai ayant entraîné une issue grave non fatale chez un sujet d’essai lors d’un essai clinique. Ces rapports doivent être envoyés au promoteur de l’essai et à l’organisme de réglementation compétent.
En résumé, la pharmacovigilance des essais cliniques implique la collecte et le signalement de différents types d’événements indésirables, d’incidents graves et d’incidents graves imputables au médicament d’essai. Ces rapports sont envoyés au promoteur de l’essai et à l’organisme de réglementation compétent afin de suivre l’innocuité des médicaments en essai et de prendre les mesures nécessaires pour protéger la sécurité des sujets d’essai.
Quelles sont les différences entre les rapports (DSURs, PSUR/PBRER, plan de gestion du risque) ?
Les rapports DSURs, PSUR/PBRER et plan de gestion du risque sont tous des outils utilisés en pharmacovigilance pour évaluer les données sur la sécurité et l’efficacité d’un médicament. Cependant, ils ont des objectifs et des contenus différents :
- Les rapports DSURs (rapports de suivi des données sur la sécurité des médicaments) sont des rapports périodiques qui rassemblent les données sur la sécurité d’un médicament au cours de son cycle de vie. Ils sont élaborés par les titulaires de l’autorisation de mise sur le marché (AMM) et envoyés aux autorités de santé réglementaires afin de leur permettre de suivre l’innocuité du médicament une fois qu’il est sur le marché.
- Les rapports PSUR/PBRER (rapports périodiques sur l’état de la sécurité/rapport périodique sur la sécurité du médicament) sont des rapports périodiques qui évaluent les données sur la sécurité d’un médicament au cours de son cycle de vie. Ils sont élaborés par les titulaires de l’AMM et envoyés aux autorités de santé réglementaires afin de les aider à évaluer l’innocuité du médicament et à prendre les décisions de réglementation appropriées.
- Les plans de gestion du risque sont des documents qui décrivent les mesures qui seront mises en place pour gérer les risques liés à un médicament. Ils peuvent inclure des plans de gestion des risques pour la sécurité des patients, les risques liés aux interactions médicamenteuses, etc. Les plans de gestion du risque sont élaborés par les titulaires de l’AMM et soumis aux autorités de santé réglementaires avant l’autorisation de mise sur le marché du médicament.
En résumé, les rapports DSURs, PSUR/PBRER et plans de gestion du risque sont tous des outils utilisés en pharmacovigilance pour évaluer les données sur la sécurité du médicament
Quels objectifs pédagogiques sont liés aux RMR/MSL ?
Le module vise à connaître la réglementation et les bonnes pratiques des essais cliniques, à comprendre le suivi de la pharmacovigilance et à cerner l’implication du RMR.
Nos thématiques de formations en ligne dédiées aux industries pharmaceutiques
Pour répondre aux exigences du secteur, IFIS Interactive vous propose des formations e-learning en phase avec l’évolution de la réglementation ainsi que de la demande des industriels.
Notre équipe est à votre disposition.
Vous souhaitez former facilement et rapidement vos collaborateurs ? Vous cherchez une solution de certification simple et innovante ? Contactez-nous ! Nos experts vous apporteront des réponses adaptées à vos problématiques.
Vous voulez également personnaliser une formation ou créer une formation sur-mesure, nos équipes sont à votre écoute.

Christel BATY

François MEGE