


Le marquage CE dispositif médical est une certification obligatoire pour permettre aux fabricants de commercialiser leurs produits dans l’espace économique européen. Nous allons donc, dans un premier temps, définir ce qu’est le marquage CE et un dispositif médical, puis les étapes, critères, et réglementations liés au marquage CE des dispositifs médicaux.
Le marquage CE dispositif médical : de quoi s’agit-il?
Le marquage CE dispositif médical est une certification de « conformité européenne » dont l’aspect est crucial dans le domaine des dispositifs médicaux. Ce marquage CE pour les dispositifs médicaux est obligatoire afin qu’ils puissent être commercialisés dans l’espace économique européen, que ce soit au sein de l’union européenne ou d’autres pays européens (Norvège ou Islande par exemple).
Le marquage CE dispositif médical a pour but de mettre en avant l’humain et sa sécurité. Les directives et règlements européens permettent, avec l’obtention obligatoire du marquage CE dispositif médical, de répondre aux besoins et exigences essentielles de sécurité, de performance, et de qualité liés au marché des dispositifs médicaux (matériovigilance).

Le marquage CE pour les dispositifs médicaux n’est pas seulement une certification de conformité, mais aussi un symbole de confiance qui a pour but de rassurer les utilisateurs et professionnels de la santé en montrant que le produit a bien été analysé de façon objective sur sa sécurité et sa performance. Le marquage CE permet, à terme, de garantir la conformité du produit afin qu’il puisse être commercialisé librement au sein du marché unique européen sans nécessiter des tests ou des certifications supplémentaires par chaque état membre.
Le marquage CE des dispositifs médicaux n’a pas pour unique vocation de certifier le produit pour sa commercialisation, cela demande également un suivi post-commercialisation où les fabricants sont tenus de surveiller la performance de leurs dispositifs médicaux après leur mise sur le marché et de signaler tout incident ou problème de sécurité aux autorités compétentes (l’ANSM pour la France par exemple) qui peuvent, par la suite, prendre des mesures correctives si cela s’avérait nécessaire comme des rappels de produits, des modifications des directives d’utilisation, ou des actions réglementaires pour assurer la sécurité des patients.
Définition des DM (dispositifs médicaux)

Selon la réglementation européenne, un dispositif médical est tout instrument, appareil, équipement, matière, produit (à l’exception des produits d’origine humaine), y compris les accessoires et logiciels, utilisé seul ou en association, à des fins médicales chez l’homme, et dont l’action principale voulue n’est pas obtenue par des moyens pharmacologiques, immunologiques ou métaboliques.. Ces dispositifs ont pour but de diagnostiquer, prévenir, surveiller, traiter ou atténuer une maladie ou une blessure, de soutenir ou de remplacer une structure anatomique ou un processus physiologique, ou de contrôler la conception. Il ne faut pas les confondre avec les médicaments car eux sont définis comme étant toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou chez l’animal ou pouvant leur être administrée, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique.
On peut citer comme exemples de dispositifs médicaux les fauteuils roulants, les lunettes correctrices, les appareils d’échographie, les couronnes dentaires, les préservatifs, les implants mammaires, les prothèses de hanche ou les seringues pour ne citer que ceux-là.
En ce qui concerne la classification des dispositifs médicaux, ces derniers sont divisés en 4 catégories dont le dénominateur commun est le degré de risque lié au dispositif pour l’Homme :
- Classe I, il s’agit de la classe de risque la plus faible (béquilles, lunettes, etc.)
- Classe IIa, il y a alors un risque potentiel modéré/mesuré (lentilles de contact, appareils auditifs, échographes, etc.)
- Classe IIb, là le risque potentiel est élevé/important (les préservatifs, appareils IRM, pompes à perfusion, sutures non absorbables, etc.)
- Classe III, la classe de risque la plus élevée (pacemaker, implants mammaires, stents coronaires, implants de hanche, etc.)
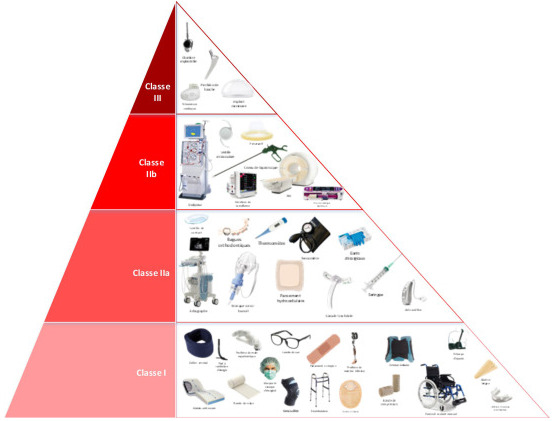
Les dispositifs médicaux sont des outils essentiels dans le secteur de la santé moderne ; en effet ils jouent un rôle clé dans la prévention, le diagnostic et le traitement des maladies. Leur développement et leur régulation rigoureux assurent que ces dispositifs médicaux peuvent être utilisé en toute confiance par les professionnels de santé et les patients. En fonction de la classification, les critères pour le marquage CE dispositif médical s’adapte afin de répondre au besoin de sécurité pour l’Homme.
Marquage CE dispositif médical : les étapes et critères pour l’obtenir
Comme vu précédemment, l’obtention du marquage CE dispositif médical implique un processus rigoureux d’évaluation de la conformité. Ce processus peut varier en complexité selon la classe de risque pour le dispositif médical. En effet chaque classe nécessite des procédures de conformité spécifiques. Ce processus peut inclure des évaluations cliniques, des tests de sécurité, et des vérifications de qualité pour garantir que le dispositif est sûr et efficace pour l’usage prévu.
Pour obtenir le marquage CE dispositif médical, la classe I (voir exemples précédemment cités) doit répondre à 5 critères :
1/ L’autocertification (les fabricants peuvent évaluer la conformité eux-mêmes sans l’intervention d’un organisme notifié).
2/ La documentation technique (obligation de fournir une documentation détaillant la conception, la fabrication et l’utilisation prévue).
3/ L’évaluation clinique (preuve de la sécurité et des performances via des données cliniques).
4/ La déclaration de conformité CE (les fabricants déclarent que le produit répond aux exigences essentielles de la directive européenne sur les dispositifs médicaux)
5/ Gestion des risques (identification et atténuation des risques potentiels liés à l’utilisation du dispositif)
Pour obtenir le marquage CE des dispositifs médicaux, la classe IIa (voir exemples précédemment cités) doit répondre à 4 critères :
1/ Evaluation par un organisme notifié (évaluation de la conformité par un organisme tiers pour certaines étapes de la conception et de la production)
2/ Documentation technique détaillée (inclure des données sur les matériaux utilisés, les processus de fabrication et les tests effectués)
3/ Evaluation cliniques et tests précliniques (preuve étendue de la sécurité et de la performance clinique)
4/ Audit de qualité (audit des systèmes de gestion de la qualité par un organisme notifié)
Afin d’obtenir le marquage CE dispositif médical, la classe IIb (voir exemples précédemment cités) doit répondre à 4 critères également :
1/ Evaluation exhaustive par un organisme notifié (évaluation détaillée des dispositifs pour l’ensemble des processus de conception et de production)
2/ Exigences de performance et sécurité rigoureuses (test clinique approfondi pour démontrer la performance et la sécurité sur une durée prolongée)
3/ Audit de la chaîne de production (audit complet de l’ensemble de la chaîne de production par un organisme notifié)
4/ Gestion des risques accrue (analyse et gestion des risques plus stricte en raison du risque plus élevé des dispositifs)
Pour finir, le marquage CE dispositif médical est attribué aux classes III s’ils répondent aux critères suivants :
1/ Examen complet par un organisme notifié (contrôle rigoureux de chaque aspect du dispositif, de la conception à la production finale)
2/ Etudes cliniques approfondies (données cliniques robustes requises pour prouver la sécurité et l’efficacité, souvent avec des essais cliniques humains)
3/ Documentation technique exhaustive (documentation détaillant tous les aspects techniques et cliniques du dispositif)
4/ Audit rigoureux des systèmes de qualité (audit fréquent des systèmes de gestion de la qualité pour garantir une fabrication constante et conforme)
5/ Suivi post-commercialisation (surveillance continue après la mise sur le marché pour identifier et corriger rapidement les éventuels problèmes de sécurité)
Les critères fixés par la commission européenne afin d’obtenir le marquage CE dispositif médical permettent de certifier que les dispositifs médicaux, en fonction de leur niveau de risque, sont sécurisés, encadrés et efficaces avant d’être commercialisés dans l’Espace économique européen.
Marquage CE dispositif médical : les réglementations

Le marquage CE pour les dispositifs médicaux est principalement régi par deux cadres réglementaires répondant aux directives et règlements européens :
1/ Le règlement (UE) 2017/745 relatif aux dispositifs médicaux (MDR) : ce règlement a remplacé les anciennes directives 93/42/CEE et 90/385/CEE et 90/385/CEE. Il établit des exigences strictes pour la conception, la fabrication et la commercialisation des dispositifs médicaux en Europe. Il s’applique à une large gamme de dispositifs, des équipements diagnostiques aux implants chirurgicaux.
La mise en place de ce nouveau règlement européen pour le marquage CE du dispositif médical amène de nouvelles évolutions réglementaires, que ce soit avant la mise sur le marché, pour l’évaluation clinique, pour l’évaluation de la conformité et les organismes notifiés, pour la mise sur le marché et la commercialisation, pour la transparence et la traçabilité (identifiant unique pour chaque dispositif, et mise en œuvre d’Eudamed par exemple) et l’harmonisation européenne.
2/ Le règlement (UE) 2017/746 relatif aux dispositifs médicaux de diagnostic in vitro (IVDR) : ce règlement concerne spécifiquement les dispositifs utilisés pour tester des échantillons in vitro, comme les kits de test de laboratoire pour les maladies infectieuses ou les conditions génétiques.
Marquage CE dispositif médical : plus qu’un indicateur, un engagement!
En conclusion le marquage CE des dispositifs médicaux permets de placer l’Homme au premier plan car tout repose sur sa sécurité et la qualité des dispositifs pour lui. Il ne s’agit pas seulement d’un indicateur de conformité mais bien d’une promesse de sécurité et de qualité pour les clients et les professionnels de santé. Le marquage CE du dispositif médical assure que les dispositifs commercialisés en Europe sont rigoureusement évalués et surveillés, garantissant à la fois la sécurité du patient et la fiabilité des dispositifs médicaux utilisés dans le cadre des soins de santé.
Il est primordial pour le fabricant de bien comprendre les réglementations associées au marquage CE afin de se conformer aux normes européennes tout en gagnant la confiance des professionnels de santé et des patients dans l’espace économique européen.
Découvrez les formations d’Ifis Interactive, spécialiste de la formation e-learning des industries de santé, pour vous former ou former vos collaborateurs à l’univers du DM:
Bonnes Pratiques Cliniques – Dispositf Médical – elearning | Ifis Interactive
Formation Dispositif Médical en e-learning | IFIS Interactive
Evaluation, Formation Charte qualité des pratiques professionnelles (ifis.fr)
La Charte de Qualité des Dispositifs Médicaux | IFIS Interactive
Avantages des formations e-learning
L’e-learning offre de nombreux avantages pour les apprenants, tels que la flexibilité, l’accessibilité et la possibilité de revenir sur des points importants à tout moment. Les formations en ligne proposent également des outils interactifs, tels que des quiz d’évaluation, pour vérifier les connaissances acquises et obtenir une attestation d’assiduité.
