


Qu’est ce que la Matériovigilance ?
La matériovigilance est un système qui vise à détecter, évaluer et signaler les effets indésirables des Dispositifs Médicaux (DM). Elle est mise en place pour améliorer la sécurité des patients en recueillant des données sur les effets indésirables potentiels des DM.
Ce système a pour objectif d’utiliser ces informations pour informer les décisions relatives à la conception, à la fabrication et à l’utilisation des DM. En effet, en recueillant des données sur les effets indésirables potentiels des DM, la matériovigilance permet de prendre des décisions éclairées pour améliorer la sécurité des patients.
Dans quel contexte s’inscrit la matériovigilance ?
Le contexte de la Matériovigilance (MV)
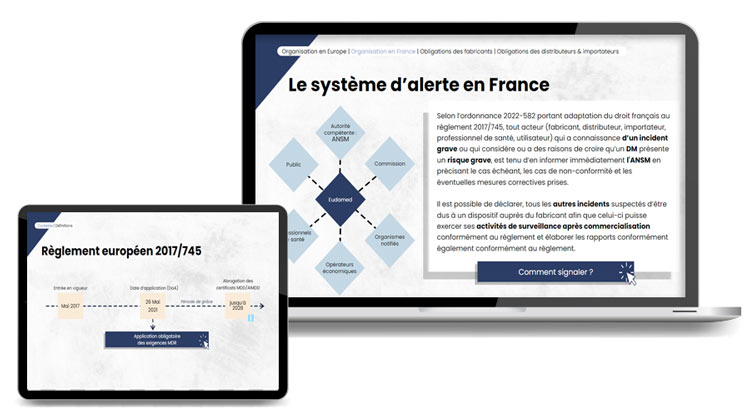
Quelques dates clés …
Depuis le 26 mai 2021, pour commercialiser un Dispositif Médical (DM) en Europe, il est obligatoire d’obtenir un marquage CE. Pour ce faire, le DM doit être vérifié pour sa conformité aux exigences de la réglementation européenne en vigueur.
Cette réglementation européenne a été mise à jour en mai 2017 avec la publication du règlement 2017/745, dénommé MDR (Medical Device Regulation). Ce règlement s’applique désormais à tout DM et prévoit une période de grâce jusqu’en 2028 pour les DM déjà commercialisés qui sont conformes aux anciennes directives européennes.
Pendant cette période de grâce, certains DM conformes aux anciennes directives européennes pourront toujours être commercialisés en Union Européenne sous certaines conditions. Cependant, depuis le 26 mai 2021, il est impératif de se conformer aux exigences du règlement 2017/745 ou MDR. Ce dernier concerne la matériovigilance et la surveillance après commercialisation. Par conséquent, l’application obligatoire des exigences MDR inclut la surveillance après commercialisation** et la vigilance, les investigations cliniques des événements indésirables, l’enregistrement des opérateurs économiques EUDAMED, ainsi que la surveillance du marché.
Le MDR exige davantage de matériovigilance et de surveillance après commercialisation. Ces exigences s’appliquent à tout DM, peu importe sa classe***, pour garantir la sécurité des utilisateurs et détecter toute anomalie ou incident lié au DM.
La surveillance après commercialisation
Les fabricants et les autres opérateurs économiques sont responsables de surveiller les DM après leur commercialisation. L’objectif est de repérer toute nécessité d’appliquer une mesure préventive ou corrective.
Il existe deux types de surveillance après commercialisation :
- La « proactive », comprenant notamment la PMCF (Surveillance clinique après commercialisation) et la surveillance de la littérature
- La « réactive », comprenant les non-conformités, les déviations et les réclamations, ainsi que la matériovigilance.
En conclusion, pour commercialiser un DM en Europe, il est obligatoire d’obtenir un marquage CE et de respecter les exigences du MDR en matière de matériovigilance et de surveillance après commercialisation. Les fabricants et les opérateurs économiques ont la responsabilité de surveiller les DM pour garantir la sécurité des utilisateurs et détecter tout incident lié au DM.
Quelques définitions sur la Matériovigilance
La matériovigilance, la surveillance après commercialisation et la surveillance du marché
La matériovigilance et la surveillance après commercialisation sont des activités cruciales pour la sécurité des dispositifs médicaux. Elles permettent de collecter des données sur leur utilisation et de détecter tout incident nécessitant des mesures préventives ou correctives.
La surveillance après commercialisation est une procédure systématique de collecte proactive de données sur les DM. Elle vise à dresser le bilan de leur utilisation et à repérer tout incident grave ou mesure de sécurité nécessaire. La surveillance du marché quant à elle consiste à vérifier que les DM sont conformes aux exigences de la législation en vigueur et ne compromettent pas la santé, la sécurité ni tout autre aspect de la protection de la santé public.
La matériovigilance a pour objectif de limiter, surveiller et détecter les incidents, effets indésirables, mésusages, défaillances ou effets néfastes des DM. Elle permet également de prendre des mesures suite aux incidents en fonction de leur fréquence et de leur gravité.
Les incidents
Un incident grave est défini comme tout incident ayant entraîné directement ou indirectement, ou susceptible d’avoir entraîné ou susceptible d’entraîner la mort d’une personne, une grave dégradation de l’état de santé ou une menace grave pour la santé publique. Une menace grave pour la santé publique est définie comme un risque imminent de mort, de grave détérioration de l’état de santé ou de maladie grave pouvant nécessiter une mesure corrective rapide.
Un incident est quant à lui défini comme tout dysfonctionnement ou altération des caractéristiques ou des performances d’un DM, y compris une erreur d’utilisation due à des caractéristiques ergonomiques, ainsi que tout défaut dans les informations fournies par le fabricant et tout effet secondaire indésirable.
Pour résumer …
En somme, la matériovigilance et la surveillance après commercialisation sont des activités essentielles pour garantir la sécurité des DM et protéger la santé publique. Les fabricants et les opérateurs économiques ont la responsabilité de surveiller les DM après leur commercialisation, pour détecter tout incident grave ou menace pour la santé publique et prendre les mesures appropriées.
Programme de la formation en ligne matériovigilance – elearning
* Un dispositif médical est un produit médical utilisé pour des fins médicales, qui n’agit pas de manière pharmacologique ou immunologique, mais peut être assisté par de tels moyens. Cela inclut les logiciels de diagnostic ou de thérapie. Les dispositifs médicaux implantables actifs sont ceux qui nécessitent une source d’énergie pour fonctionner. La définition des dispositifs médicaux est identique dans tous les pays de l’Union européenne.
Les dispositifs médicaux sont classés en quatre catégories en fonction de leur risque potentiel pour la santé, avec des réglementations différentes pour chaque catégorie : Classe I (faible risque), Classe IIa (risque modéré), Classe IIb (risque élevé), Classe III (risque très élevé). La classification est effectuée par le fabricant en utilisant les règles de la directive sur les dispositifs médicaux, en fonction de la finalité médicale revendiquée pour son produit.
** Chapitre VII, section 2
*** 26 mai 2026 : Classe II implantables sur mesure
31 décembre 2027 : classes IIb et III implantables hors sutures, agrafes, produits d’obturation dentaire, appareils orthodontiques, couronnes dentaires, vis, cales, plaques, guides, broches, clips et dispositifs de connexion
31 décembre 2028 : autres DM impliquant un organisme notifié pour le règlement

Eric Villemagne, Directeur Général chez IFIS Interactive depuis plus de 10 ans, incarne avec excellence l’expertise et l’innovation dans le domaine de la formation e-learning pour les industries de santé. Fort d’une ancienneté de plus de 20 ans au sein du groupe IFIS et d’une expérience enrichissante de 25 ans dans le secteur de la formation, sa carrière est marquée par une contribution significative à l’évolution et à l’amélioration des processus d’apprentissage. Diplômé d’un Executive MBA de l’EM Lyon, Eric s’engage avec détermination à fournir des formations sur étagère et sur mesure, répondant précisément aux besoins des professionnels de l’industrie pharmaceutique, du dispositif médical et de la cosmétique.
Sa vision stratégique ne s’arrête pas là. Eric ambitionne de contribuer activement à la relocalisation et au développement durable de ces industries cruciales en France et en Europe. Par ce biais, il affirme non seulement son leadership mais également son engagement indéfectible envers l’excellence éducative et industrielle. Sa démarche vise à instaurer un environnement propice à l’innovation et à la croissance durable, en mettant l’accent sur des solutions de formation qui anticipent et répondent aux défis de demain.
